2月18日,北京大学生物医学前沿创新中心(BIOPIC)、生命科学学院、北京未来基因诊断高精尖创新中心(ICG)、生命科学联合中心(CLS)张泽民实验室联合百奥智汇在期刊《Genome Biology》上发表了题为 “iMAP: integration of multiple single-cell datasets by adversarial paired transfer networks”的生物信息方法学论文,提出了基于深度自编码器和生成式对抗神经网络的单细胞数据整合的新方法iMAP。

整合多来源的数据集是利用单细胞RNA测序技术产生可靠的新发现的重要途径。然而,不同批次实验产生的数据集之间存在不可避免的技术差异,消除这些技术差异而保留不同实验之间真实存在的生物学差异,是开发批量效应消除方法的主要挑战。目前的主流批次效应消除方法都很难在两者之间做到可靠的平衡。
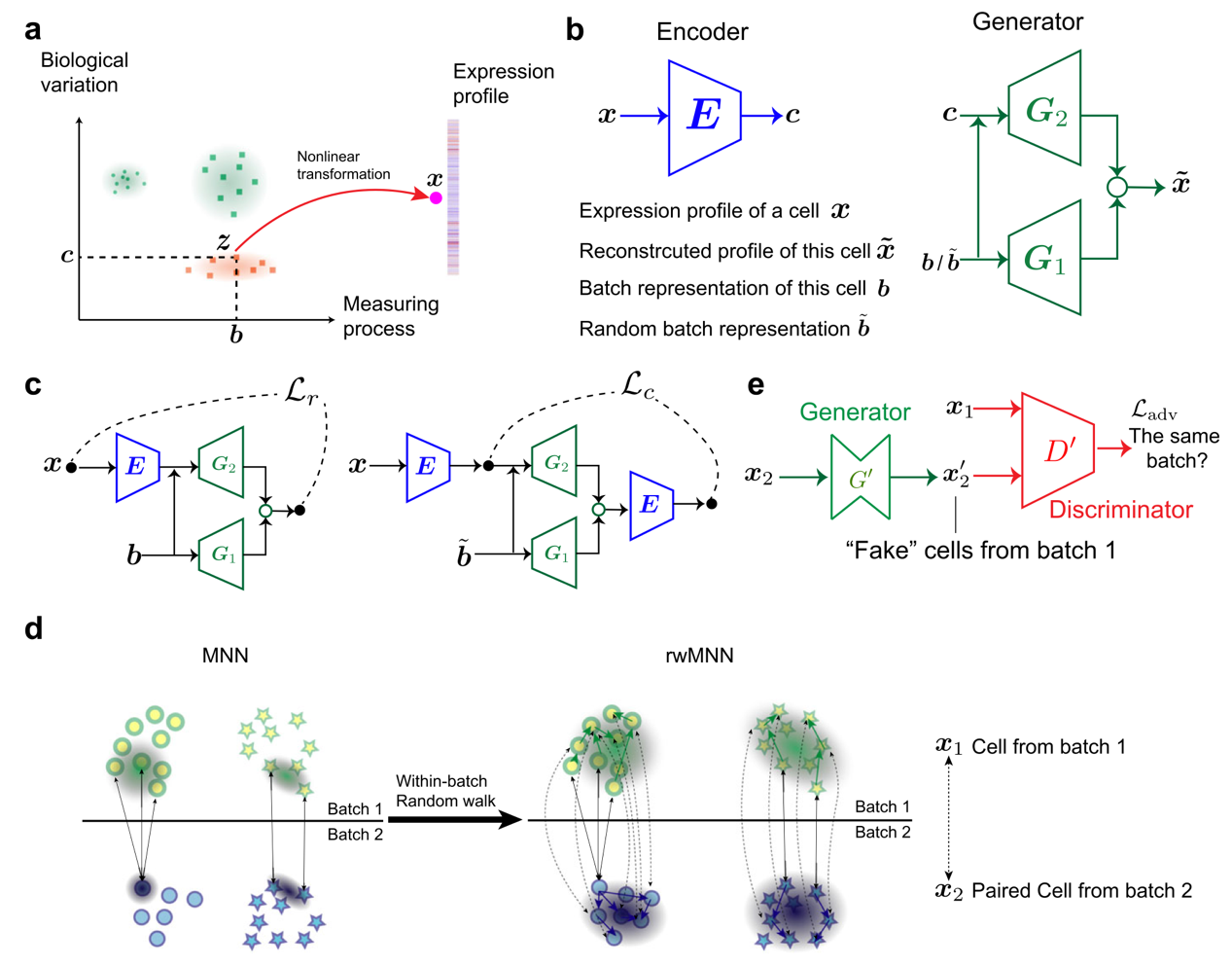
张泽民实验室的博士后王东方等开发了一种新的方法iMAP,为单细胞数据的有效整合提供了新的思路。他们开发的iMAP方法结合了目前两种最先进的无监督深度网络结构—深度自编码器和生成式对抗神经网络(GAN)(图1)的优势。GAN的主要作用在于能够准确地将不同数据集、相同细胞类型的细胞的基因表达分布进行混合,然而真实生物数据集的细胞组成十分复杂,可能存在不完全重合的细胞类型,相同细胞类型在不同数据集中的分布比例也可能存在很大差异。因此,iMAP首先构建了一种新的自编码器结构来提取细胞的低维表示特征,该特征能够一定程度上消弭批次效应的影响,同时保留不同数据集之间真实存在的生物学差异,进而通过构建rwMNN细胞对,形成有效的自训练数据指导后续GAN网络进行正确的细胞基因表达分布混合。与其他方法相比,iMAP既能匹配不同批次数据集中相同类型的细胞的基因表达分布,又能识别各个数据集上特定的细胞类型。他们在十多个不同规模、不同测序技术产生的数据集上论证了iMAP方法的有效性与可靠性。与其他基于深度学习的方法相比,iMAP在大规模数据集上具有显著的速度优势。他们也将iMAP应用于肿瘤浸润免疫细胞数据集的分析,通过整合分别由Smart-seq2和10x Genomics技术产生的数据集发现了肿瘤微环境中新的细胞间相互作用。
iMAP提供了免费Python软件包 (https://github.com/Svvord/iMAP),可供用户实现单细胞转录组数据整合。随着单细胞测序技术的广泛普及应用以及大量的大规模数据集的产生,iMAP可能成为整合不同批次实验产生的数据的有利工具,并为后续算法的开发提供新的思路。
北京大学BIOPIC/生命科学学院博士后王东方和清华大学博士生侯思宇为该论文的共同第一作者,王东方和BIOPIC/生命科学学院张泽民教授为该论文的通讯作者。该课题得到了国家自然科学基金委、北京未来基因诊断高精尖创新中心及北京百奥智汇的资助。